GIBH Develops a Downstream Analysis and Visualization Tool for Quantitative Proteomic Data
Quantitative proteomics based on high-performance liquid chromatography-mass spectrometry (HPLC-MS) has emerged as an important tool in life science research. Like other omics methodologies, a well-defined and systematic data analysis workflow is essential for effective research. However, the diverse instrumentation and quantitative software used in mass spectrometry pose challenges for downstream data processing and analysis of acquired proteomic data. Consequently, there is an urgent demand for a downstream analysis tool capable of effectively handling the diverse nature of proteomic data.
The team, led by Dr. ZHANG Xiaofei from the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, recently published a paper entitled "DEP2: An Upgraded Comprehensive Analysis Toolkit for Quantitative Proteomics Data"in Bioinformatics. The paper presents DEP2, an R package capable of processing input data from multiple quantitative proteomics software.
Utilizing S4 objects for data packaging and implementing the limma package for differential analysis, DEP2 serves as a versatile tool for downstream data processing, differential analysis, and visualization in quantitative proteomics. DEP2 integrates algorithms for peptide-to-protein re-quantification and missing value imputation. It enables the analysis of peptide-level and protein-level quantitative proteomics, as well as modification-specific proteomics data.
Additionally, DEP2 features a modular Shiny application that enables simultaneous processing of multiple proteomics data without the requirement for coding.
DEP2 is compatible with various types of upstream results, providing convenience for researchers in handling proteomic data. The open-source R packaged is available at https://github.com/mildpiggy/DEP2.
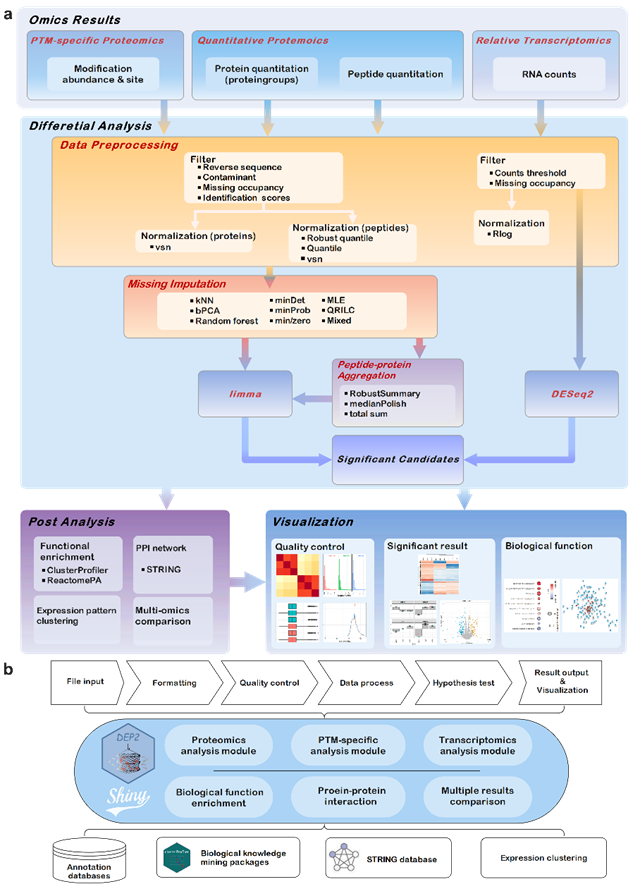
Fig.1 Schematic overviews of DEP2 analyses and built-in application (Image by GIBH)