GIBH’s new progress of treating clinically non-small-cell Lung cancer (NSCLC) patients with acquired resistant
Lung cancer is the leading cause of cancer deaths in the worldwide. Non-small-cell lung cancer (NSCLC) accounts for about 85% of all lung cancers.
Small molecular inhibitors of epidermal growth factor receptor (EGFR), gefinitib and erlotinib, have achieved significant clinical benefit for NSCLC patients. But their efficacy is eventually diminished because of acquired resistance to them. Particularly, a single T790M point mutation (threonine790→methionine790) at the “gatekeeper” position in EGFR accounts for approximately 50% in clinically acquired resistant patients. Although the second-generation Cys797-chelating irreversible EGFR inhibitors such as canertinib, afatinib, neratinib and pelitinib displayed promising potential to overcome EGFRT790M related resistance in animal models,their non-selective inhibition against wild-type EGFR (EGFRWT) and/or other kinases results in a relatively low maximal-tolerated-dose (MTD) and poor clinical outcomes in human patients.
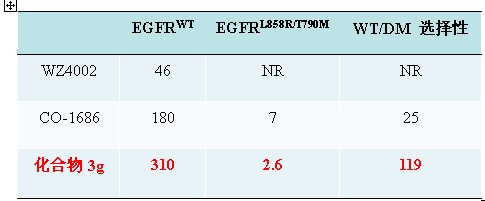
Inhibitors selectively targeting EGFRT790M mutants represent a novel attractive strategy for the clinical management of NSCLC patients with acquired resistance. Gray et al. have reported the first novel class of substituted pyrimidines as irreversible EGFR inhibitors in 2009. Their representative compound WZ4002 showed good selectivity against EGFR T790M mutants over the wild type kinase. Clovis Oncology started a Phase I study of their mutant-selective EGFR inhibitor CO-1686 in 2012. CO-1686 whose chemical structure was not disclosed showed only about 25-fold [Kd(EGFRWT ) / Kd(EGFRL858R/T790M) = 25] selectivity.
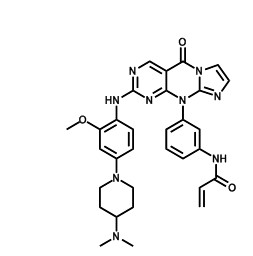
Prof. Ke Ding’s group at GIBH has designed and synthesized a series ofpyrimido[4,5-d]pyrimidin-4(1H)-one based EGFRT790M inhibitors with >100- fold selectivity over the wild-type kinase. The representative compound 3g binds to EGFRL858R/T790M and EGFRT790M with Kd values of 2.6 and 1.3nM, respectively and it is 120-240 times less potent with respect to wild-type EGFR. It also strongly and selectively inhibits the EGFR signal transduction and proliferation of H1975 NSCLC cells harboring the EGFRL858R/T790M mutation(IC50=86nM), but were significantly less potent with NSCLC or normal cells with wild-type EGFR. In a panel of 456 kinases, 3g displayed remarkable selectivity and is one of the most selective EGFR T790M inhibitors reported to date. Parts of the group research results were published in Journal of Medicinal Chemistry in May 2013 (2013, 56,4738–4748) and Angewandte Chemie International Edition in June 2013 ( DOI: 10.1002/anie.201302313).
Contributor:Tianfeng Xu Chemical Biology Institute